The quality of newly generated study data is a decisive factor after resubmission due to new scientific findings as well.
In the Rules of Procedure (VerfO) Verfahrensordnung (g-ba.de) of the Federal Joint Committee (G-BA) the requirements for a dossier due to new scientific findings are specified in Chapter 5 Section 13 of the German Social Code, Book Five (SGB V). The G-BA can decide based on the new scientific findings on a new benefit assessment of a drug to be evaluated at the request of its members, or of the organizations and institutions specified in Section 139b (1) sentence 2 SGB V. The earliest time that the evaluation can take place is one year after the decision on a benefit assessment according to Section 20. This also applies if the therapeutic indication of the drug has been restricted by the responsible regulatory authorities. In addition to this, it is also stipulated that the pharmaceutical company is to be offered consultation in accordance with Section 7.
To date, the G-BA has conducted nine procedures under Section 13 ( as of December 2022) − among these, within three procedures (33.3%) the study data were consulted by the G-BA for an assessment of new scientific evidence, three pharmaceutical companies did not present the study data in the submitted dossier (33.3%), once (11.1%) the pharmaceutical company did not submit a dossier, and two studies (22.2%) were classified by the G-BA as not suitable for deriving an additional benefit.
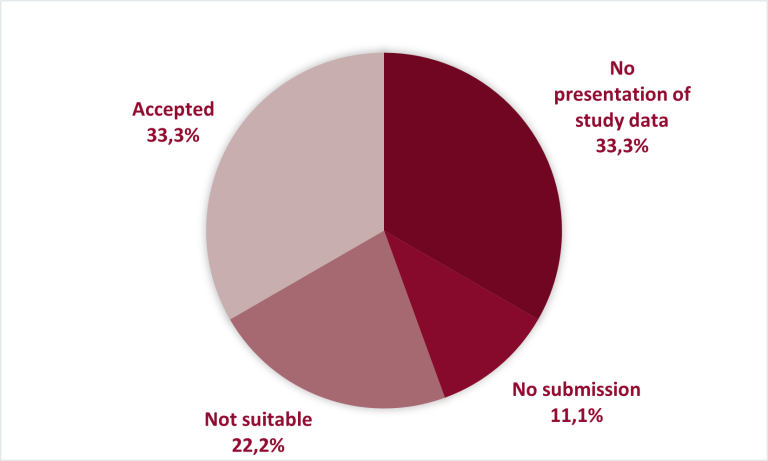
Figure 1: G-BA assessments of the available evidence
Why were studies not submitted?
In one case no dossier was submitted for a new benefit assessment because, according to the pharmaceutical company, it was methodologically not possible to present an additional benefit based on a direct comparative study due to the determined appropriate comparator and inhomogeneity of the patient population of the existing studies did not allow an indirect comparison.
In one procedure in an oncological indication, the study population investigated did not correspond to the patient populations defined by the G-BA.
At the time of the new benefit assessment, no comparative results were available for two other oncology drugs. The basis of the request by the G-BA was in each case changes in the therapeutic indication of the substances due to early signals from the studies still ongoing at that time. Concerning the cases that are not considered by the G-BA, the patient population, the determined appropriate comparator, and the implementation of the Summary of Product Characteristic (SmPC) in the clinical trials are known hurdles within the benefit assessment by the G-BA.
Why are study data with new scientific findings not considered?
The request of the members for a new benefit assessment according to Section 35a (1) SGB V in conjunction with Section 3 (1) No. 4 on the Benefit Assessment of Pharmaceuticals (AM-NutzenV) and Chapter 5 Section 13 VerfO for a substance in the therapeutic indication of musculoskeletal diseases was submitted following a review of the safety profile in the context of pharmacovigilance. According to the G-BA, this no longer represents an adequate treatment for the patients in the study relevant for the re-evaluation, considering the warnings and precautions for use after updating the SmPC.
The second case concerns an active substance in the therapeutic area of infectious diseases in 2022 in which the G-BA justifies that the study is not suitable for deriving an additional benefit and cannot be regarded as new evidence in the present area of therapeutic indication. The G-BA cites as reasons the lack of implementation of appropriate comparator and the fact that the dosage was not fully in compliance with the SmPC.
Consultation before market access
The possible outcome of a trial can be anticipated by a strategically well-planned and time-tailored approach. In this context, the request for consultation represents the first important milestone before market access and during the life cycle of each drug. A product-specific presentation of the medical evidence, extensive research on the appropriate comparator, and the specific preparation of the G-BA consultation by interdisciplinary teams are among the main factors for successful market access and continued existence on the market.
You have any questions?
Just contact us by email and we will be happy to advise you.